
Research in the St. Hilaire Lab focuses on characterizing the underlying pathobiology of vascular disease, concentrating on mechanisms that drive vascular and valvular calcification and remodeling. Ectopic calcification in the cardiovascular system was once considered a passive and degenerative progression, but it is now appreciated that active biological processes drive this pathology, though the precise mechanisms inducing and propagating pathological calcification are not completely understood. Our overarching goal is to dissect the mechanisms that drive the transformation of a healthy vascular cell into a calcifying cell, in order to identify targets for the development of pharmacological therapies.
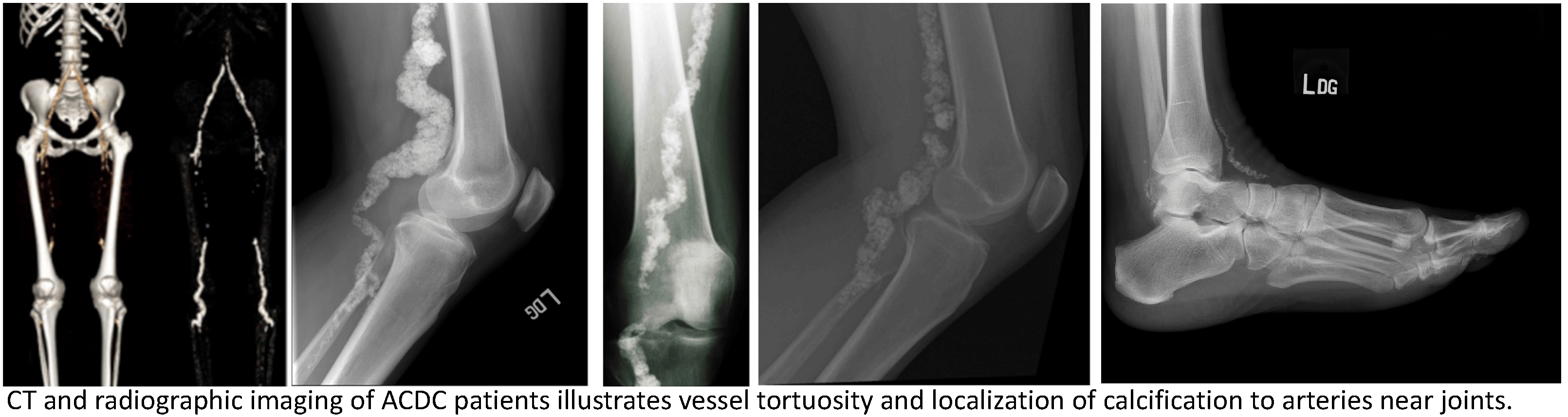
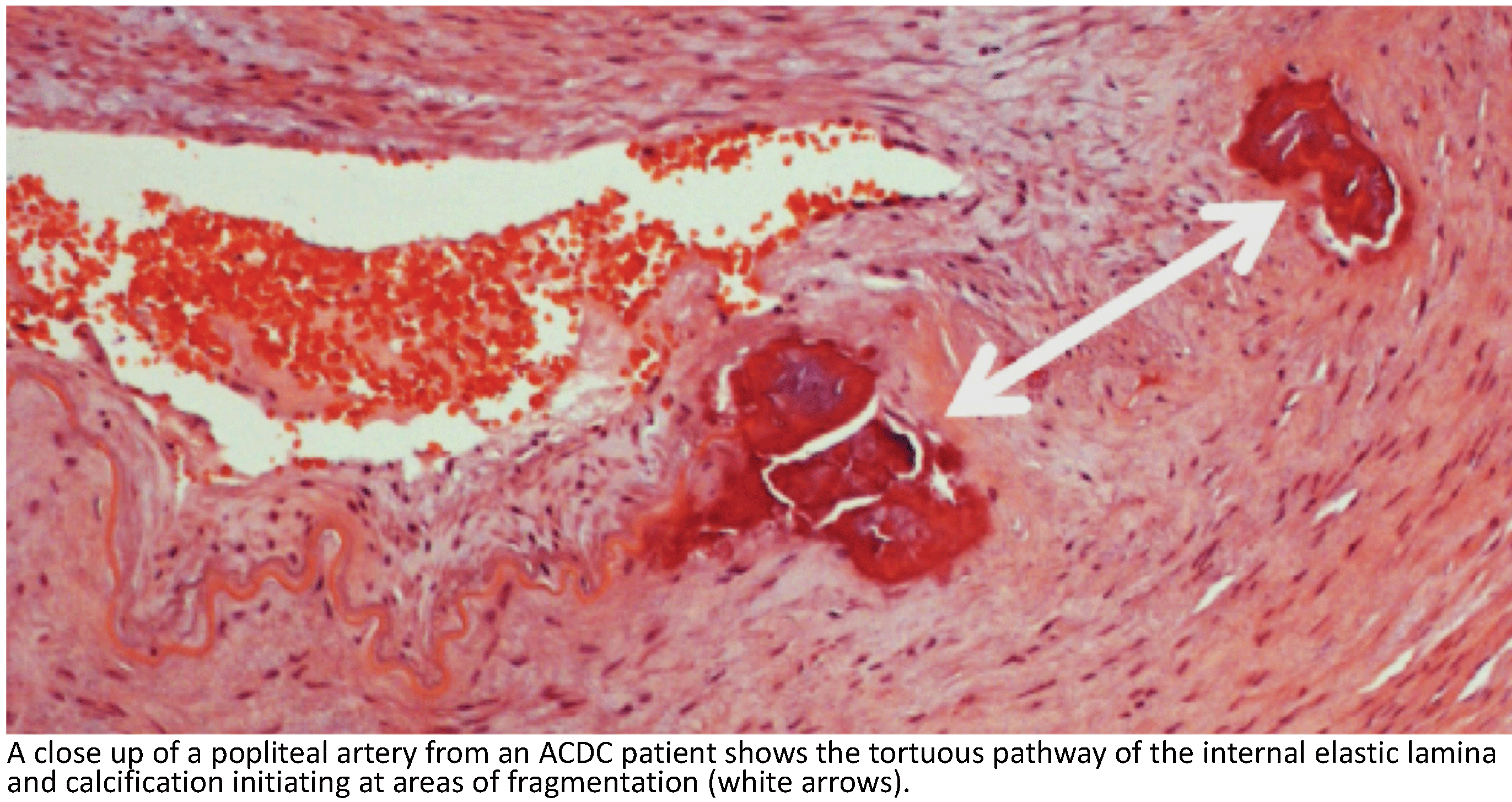
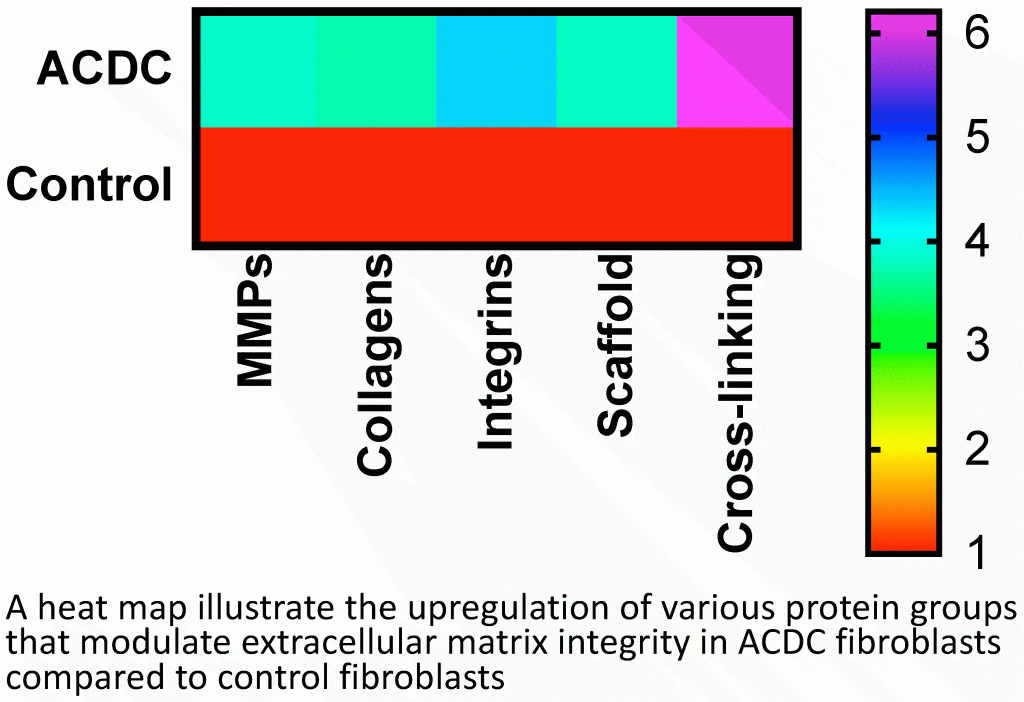
One avenue of research stems from the discovery by Dr. St. Hilaire and her colleagues at the NIH of the rare disease Arterial Calcification due to Deficiency of CD73 (ACDC). Patients with ACDC have inactivating mutations in the gene encoding for CD73, which catalyzes the breakdown of extracellular AMP to adenosine. ACDC patients exhibit severe medial-layer lower extremity vascular calcification and vessel tortuosity, which suggests that CD73 and adenosine signaling help to protect the vasculature from these pathologies. Vascular calcification and vessel tortuosity are phenotypes seen in a variety of diseases thus we aim to leverage what we learn from the study of this rare disease to identify novel mechanisms driving these pathologies in more common disorders.
Calcification in the aortic valves develops in roughly 30% of the aging population. Calcific aortic valve disease (CAVD) encompasses a spectrum of pathologic states that range from aortic sclerosis, the thickening and stiffening of the valve leaflets, to aortic stenosis, where blood flow to the aorta is obstructed and can lead to heart failure. We have created a human biospecimen bank of 150 control and CAVD valve specimens from which we obtain samples for pathology, biochemical, and gene expression analysis, as well as create primary cell lines that we use to study the process of how a healthy valve cell converts into a calcifying valve cell.
MAJOR RESEARCH QUESTIONS
1. How does lack of CD73 and adenosine signaling drive vascular calcification?
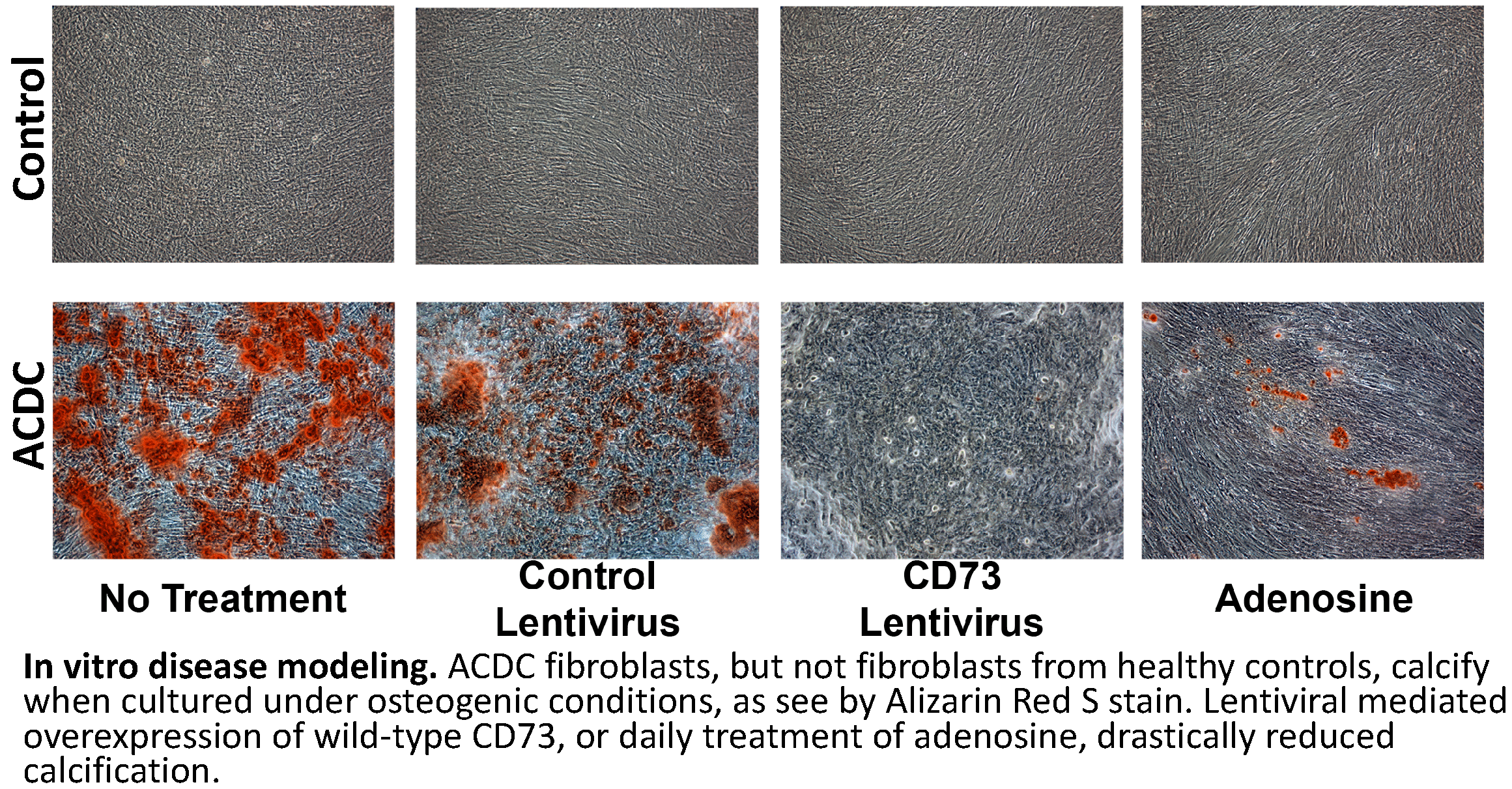
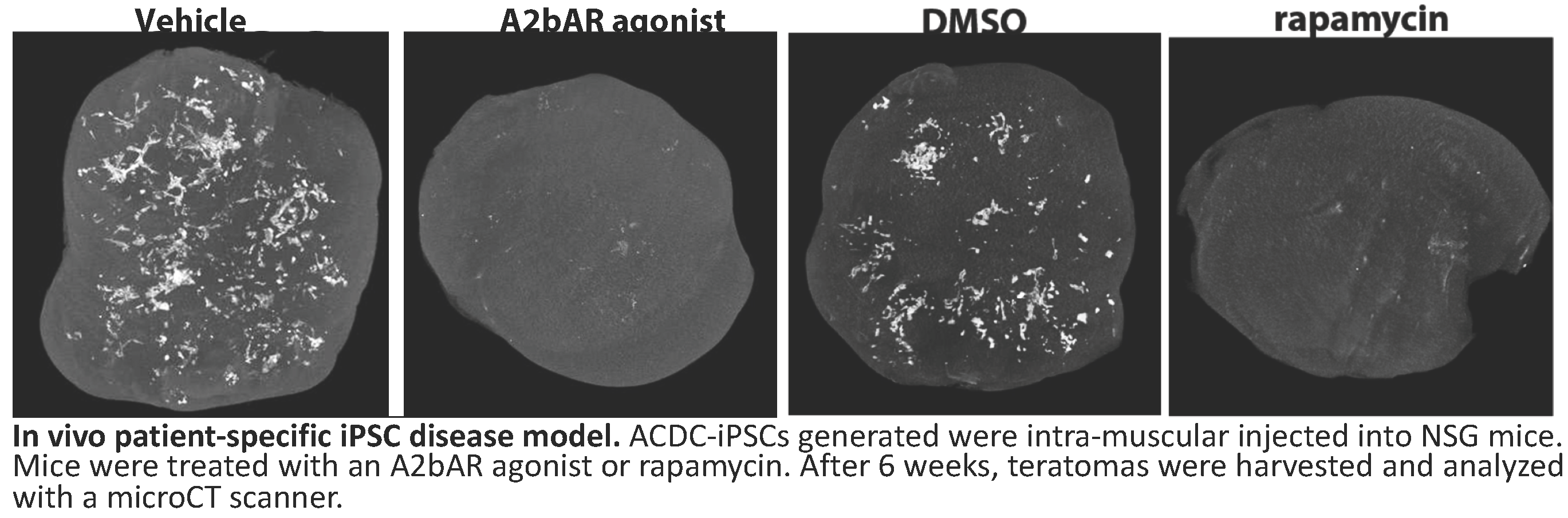
2. How does CD73 and adenosine signaling protect vessel integrity?
3. How does a healthy valve interstitial cell transition into a calcifying cell?
Calcific aortic valve disease (CAVD) is a common disorder strongly associated with aging that causes substantial medical care costs and health burden. The only current therapy is aortic valve replacement. Without replacement, mortality rates approach 50% within 2-3 years of diagnosis. Aortic valve replacement does not come without risks, as mechanical valves, while stable, require life-long anticoagulation therapy, and bioprosthetics last only a decade. Thus, there is a great need for early non-surgical therapeutic interventions. Our long-term goal is to dissect the mechanisms that transform a healthy valve into a calcifying valve, in order to identify potential targets for the development of non-surgical therapies.
Valve interstitial cells (VICs) are the predominant cell type within the valve and reside in a quiescent state, however genetic predisposition, environmental cues, inflammation, and exposure to mechanical and biochemical stresses over time disrupt VIC homeostasis. With loss of homeostasis, VICs acquire a myofibroblast-like phenotype capable of proliferation, contraction, and production and secretion of proteins that remodel the extracellular millieu. These cells, called activated VICs, alter the mechanical properties of the valve and secrete cytokines that drive activated VICs to differentiate into osteogenic calcifying VICs. Ectopic calcification that occurs in the soft tissues, such as vessels and valves, is an active biological process that shares many features with physiological bone formation and remodeling. Hallmarks of the switch from a quiescent VIC to an activated myofibroblast-like VIC include proliferation and migration of these cells, and increased production of smooth muscle alpha actin, collagens, and extracellular matrix (ECM) remodeling proteins. Gene transcription array analyses comparing healthy and calcified valves have identified the differential expression of hundreds of genes in pathways involved in inflammation, lipid retention and oxidation, ECM remodeling, angiogenesis, and calcification. While these analyses have identified numerous factors that contribute to CAVD, the early events that drive the concerted activation of osteogenic genes remain ill-defined. Our central hypothesis is that inflammatory and mechanical stress activates osteogenic gene transcription and promotes the transition of quiescent valve interstitial cells (VICs) into calcifying VICs.
![]()